科普 | 传说中的“铜娃娃”——肝豆状核变性
沈女士的宝宝刚出生3天,表现均正常,在湖北省妇幼保健院进行了新生儿遗传病基因筛查,结果提示存在肝豆状核变性患病风险,新筛中心的工作人员立即联系沈女士夫妇,告知他们尽早咨询医生,做进一步检查。
最后,夫妻双方的基因检查结果显示,沈女士夫妇均为该致病基因的携带者,而宝宝正好遗传了他们两个的致病基因,确诊为肝豆状核变性患者。那么肝豆状核变性是一种什么疾病?有哪些临床表现?又该如何预防呢?今天就让我们一起来了解一下。

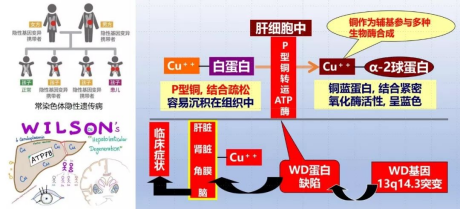
肝豆状核变性又称Wilson病(WD),属于一种常染色体隐性遗传病,由位于第13号染色体的ATP7B 基因突变导致体内铜离子转运及排泄障碍,铜在肝脏、神经系统、角膜、肾脏等脏器蓄积,出现一系列临床表现,患儿又被称为“铜娃娃”。
肝脏铜蓝蛋白合成减少,胆道铜排泄障碍,铜在肝脏沉积,肝细胞坏死,所释放的游离铜沉积于神经、肾脏、角膜等其他脏器,导致多脏器损害。
患者的父母往往无病,但各自携带一个ATP7B的致病性变异,当双亲的变异传递给同一个子女时(概率为1/4),即产生了WD患者。肝豆状核变性在人群中的发生率为1/30 000,ATP7B基因突变携带率为1/90,90个人中就有一个携带突变基因。
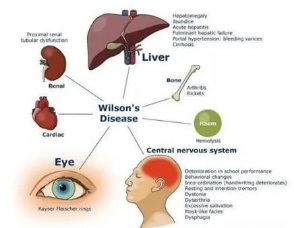
肝豆状核变性可累及全身多个脏器,临床表现多样。发病年龄多为3~60岁,也有8个月及70多岁发病的患者报道。儿童患者多以肝脏受累为首发表现,青少年及成人以神经系统受累为首发症状的患者较多。
表现为无症状的转氨酶升高,慢性肝炎,肝硬化和急性肝功能衰竭等。
运动功能障碍、震颤、共济失调、舞蹈症、自主运动障碍、肌张力障碍,一些患者表现为面具脸、四肢僵硬、步态异常等。
情感障碍,行为异常,智力减退,痴呆,性格改变。
位于角膜与巩膜交界处的K-F环,呈绿褐色或金褐色。
血尿、蛋白尿、微量蛋白尿等。
骨质疏松、病理性骨折、骨关节畸形等。
溶血性贫血、肝硬化、脾功能亢进致血液三系下降、凝血功能异常等。
肝豆状核变性患者治疗原则
减少铜摄入,阻止铜吸收,排出体内多余的铜,维持体内铜代谢平衡。早发现,早诊断,早治疗,在医生指导下终身低铜饮食和药物治疗。出现暴发性肝衰竭、失代偿性肝硬化、药物治疗无效和难以控制的神经系统症状时,可考虑肝移植。对于出现了神经、血液等系统症状的患者,可分别予对症治疗。
肝豆状核变性为常染色体隐性遗传病,患者父母再次生育再发风险为25%。应对所有患者及其家庭成员提供必要的遗传咨询,对高风险胎儿进行产前诊断。
➡➡想要获取更多针对性医疗或用药咨询,详请登录“湖北省妇幼保健院”,预约【优生遗传中心】门诊号后,来院或扫码咨询。









作者:吴倩
审核:宋婕萍
编辑制作:丰小慧
图片:部分图片来源网络